


Estudo colaborativo reúne dados sobre o mais recente surto de febre amarela no país
Mapeamento do genoma completo do vírus foi aliado à análise da origem, evolução e rotas de dispersão de casos, além da criação de modelos tridimensionais das proteínas do vírus
Um estudo colaborativo conduzido por pesquisadores de cinco laboratórios do Instituto Oswaldo Cruz (IOC/Fiocruz) reúne diversas evidências relacionadas ao vírus da febre amarela associado ao surto mais severo da doença na América do Sul nos últimos 70 anos, ocorrido entre os anos de 2017 e 2018. A iniciativa que conta com a parceria do Programa de Computação Científica (PROCC) da Fiocruz e de instituições de Minas Gerais e do Espírito Santo envolveu análises genéticas, o entendimento das rotas de dispersão dos casos e a modelagem da estrutura de proteínas do vírus. Nossos resultados sobre dimensões variadas do surto só foram possíveis por conta da parceira estabelecida entre profissionais com conhecimentos complementares. Esta união permitiu avançarmos em respostas sobre esse desafio de saúde pública, destaca a biologista molecular Myrna Bonaldo, chefe do Laboratório de Biologia Molecular de Flavivírus do IOC e uma das lideranças na investigação.
No início de 2017, em meio à primeira onda de casos, especialistas do Laboratório de Biologia Molecular de Flavivírus e do Laboratório de Mosquitos Transmissores de Hematozoários do IOC se empenhavam em buscar respostas que pudessem esclarecer se existia algo de diferente no vírus da febre amarela que estava circulando e que pudesse favorecer a rápida proliferação de casos [relembre clicando aqui].
Na ocasião, os cientistas descreveram o genoma completo de duas amostras do vírus provenientes de dois macacos bugios (espécie Alouatta clamitans) que adoeceram em uma área de mata no Espírito Santo. Os resultados, publicados na revista científica Memórias do Instituto Oswaldo Cruz, constataram a presença de variações em sequências genéticas. Estas mutações estavam associadas a proteínas envolvidas no processo de replicação viral o que tem potencial de influenciar a velocidade de propagação de casos, mas não impacta na efetividade da vacina utilizada para prevenção da doença.
|
Josué Damacena |
|

|
|
Estudo colaborativo reuniu integrantes de cinco laboratórios do IOC e de unidade da Fiocruz: empenho em dar respostas à sociedade |
Para complementar as evidências obtidas na pesquisa anterior, na busca de dados para explicar as implicações biológicas e epidemiológicas do achado inicial, era necessário, por exemplo, realizar o sequenciamento do genoma de mais vírus relacionados ao surto, tanto em casos humanos, como em mosquitos e em macacos infectados.
Com esses objetivos em mente, os especialistas concluíram o genoma completo de mais 12 amostras do vírus da febre amarela, provenientes de mosquitos, humanos e primatas infectados em 11 localidades dos estados do Rio de Janeiro e do Espírito Santo. Os dados, publicados na revista internacional Journal of General Virology, incluem duas espécies de mosquitos coletados no Espírito Santo (Haemagogus leucocelaenus e Haemagogus janthinomys, responsáveis pelo ciclo silvestre da doença), além de quatro macacos bugios e dois saguis e de quatro casos humanos, todos do Rio de Janeiro. Esta etapa foi possível a partir da colaboração da Superintendência de Vigilância Epidemiológica e Ambiental da e do Laboratório Central de Saúde Pública Noel Nutels (LACEN-RJ), ambos ligados à Secretaria Estadual de Saúde do Rio de Janeiro; do Núcleo Especial de Vigilância Ambiental da Secretaria Estadual de Saúde do Espírito Santo; e do Instituto Federal do Norte de Minas Gerais.
Assim como na análise anterior, os resultados apontam que os microrganismos possuem uma característica em comum: todos pertencem ao subtipo genético conhecido como genótipo Sul Americano 1 clade E, que é predominante nos casos de febre amarela detectados no Brasil desde 2008. Da mesma forma, uma comparação entre os 12 genomas revelou que os vírus tinham sequências genéticas praticamente idênticas. Foram identificadas, no total, um conjunto único de nove alterações de aminoácidos nas proteínas virais em todas as amostras consideradas no estudo. Estas alterações estão presentes somente nas cepas circulantes no surto recente. Nenhuma outra sequência genética depositada nos bancos de dados internacionais destinados a esta finalidade possui semelhança com as brasileiras, comenta a pesquisadora Myrna Bonaldo, que coordenou o estudo com o pesquisador Ricardo Lourenço, chefe do Laboratório de Mosquitos Transmissores de Hematozoários do Instituto. A partir dos novos dados, foi possível confirmar uma nona mutação: no primeiro estudo, já havíamos identificado oito mudanças de aminoácidos, que também foram encontradas, acrescidas de mais uma. Essas mutações se assemelham à impressão digital dos vírus. Os patógenos que estão causando o surto em nosso país são únicos, não identificados em nenhum outro local até o momento, completa Lourenço.
|
Josué Damacena |
|

|
|
Os mosquitos Haemagogus, como o Hg. leucocelaenus, são vetores primários da febre amarela silvestre |
O grupo de especialistas reforça que as mutações identificadas não têm impacto sobre a eficácia da vacina de febre amarela ofertada pelo Ministério da Saúde brasileiro. O conjunto de mutações que nosso grupo de pesquisa identificou no vírus da febre amarela em circulação no país, descrito em ambas as etapas do estudo, impacta apenas na produção de duas proteínas da estrutura do vírus, porém não afetam em nada as proteínas do envelope do vírus, que é o alvo da resposta de anticorpos neutralizantes provocada pela vacina, enfatiza Myrna. Em teoria, para um possível impacto em uma vacina, a questão é muito menos a quantidade de mutações, mas a forma como estas mutações podem influenciar a produção de proteínas. Para impactar a vacina, as mutações precisariam afetar trechos do genoma ligados à produção de proteínas do envelope, aspecto não identificado nos estudos.
A rota do vírus pelo Sudeste
Nesta segunda etapa, a parceria envolvendo cinco laboratórios do IOC também rendeu descobertas inéditas sobre a dispersão do vírus no território nacional. Especialistas do Laboratório de AIDS e Imunologia Molecular do IOC se empenharam em analisar os dados genéticos dos 14 genomas dos vírus (os dois genomas obtidos inicialmente, no início de 2017, e os 12 obtidos na fase mais recente do estudo) com o objetivo de desvendar a origem do vírus da febre amarela que está circulando no Sudeste, sua evolução e suas rotas de dispersão pelo país. Para reconstruir a dinâmica de disseminação do vírus, os pesquisadores estudaram a fundo as mutações no genoma viral e construíram uma árvore filogenética similar a uma árvore genealógica, ela agrupa no mesmo ramo os vírus que compartilham um ancestral comum.
Com base em um método probabilístico para avaliar traços hereditários, o estudo foi capaz de estimar que o vírus da febre amarela relacionado ao surto circula no Brasil pelo menos desde abril de 2016. Este achado mostra que o vírus já estava infectando mosquitos e macacos cerca de nove meses antes dos primeiros casos humanos serem confirmados e reportados ao Ministério da Saúde, em janeiro de 2017, diz Gonzalo Bello, pesquisador do Laboratório de Aids e Imunologia Molecular do IOC, que conduziu essa etapa do estudo.
Uma explicação para o início das infecções humanas haverem sido detectadas apenas meses depois é o fato de se tratar do ciclo silvestre da doença. O ser humano entra nesse ciclo de forma acidental. É necessário que uma pessoa sem imunidade entre na mata ou floresta e seja picada por um mosquito infectado, que provavelmente estava buscando uma fonte alternativa de alimentação, uma vez que os primatas da região já deveriam estar mortos, pois são muito suscetíveis ao vírus, completa Bello. O cientista já havia conduzido um amplo estudo envolvendo a análise genética dos vírus da febre amarela relacionados a casos brasileiros ao longo dos últimos 60 anos, publicado em agosto de 2017 na revista internacional Scientific Reports. Os resultados haviam apontado que os vírus relacionados ao surto recente não derivam dos vírus que causaram o surto anterior no Sudeste, entre 2008 e 2009, mas seriam resultados de uma nova introdução a partir da Venezuela (ou a partir da região Amazônica) [relembre o estudo].
|
Gutemberg Brito |
|

|
|
Os pesquisadores analisaram as mutações no genoma viral e construíram uma árvore filogenética, que apontou que a linhagem moderna do vírus da febre amarela chegou ao Brasil por dois caminhos |
Para entender melhor a dinâmica espaço-temporal da disseminação do vírus na região Sudeste, a mais afetada no surto recente, os cientistas utilizaram uma variedade de modelos filogeográficos. Os resultados indicam que o vírus chegou ao estado do Rio de Janeiro, que não reportava casos da doença havia mais de 70 anos, partindo do Espírito Santo por pelo menos três vias, entre o final de fevereiro e início de abril de 2017. Em seguida, houve uma rápida disseminação para o sul do estado, na direção da grande área metropolitana da região fluminense.
De acordo com a pesquisa, o vírus foi se dispersando dentro do estado do Rio de Janeiro seguindo duas rotas principais. Na primeira, o patógeno chegou ao município de São Fidélis (no Nordeste do estado) em fevereiro de 2017, avançando na direção sul ao longo do lado litorâneo da Serra do Mar, chegando finalmente ao município de Guapimirim, na grande região metropolitana, no início de junho de 2017. Na segunda rota, o vírus chegou ao município de Carmo, ao norte do estado, no início de abril de 2017, e seguido a rota para o sul, ao longo da parte norte ou interior da Serra do Mar, atingindo a grande região metropolitana do Rio de Janeiro no final de abril de 2017, por meio da cidade de Petrópolis.
De acordo com nossas estimativas, o vírus se espalhou do sul do Espírito Santo para a grande área metropolitana do Rio de Janeiro com uma taxa média de dispersão de 3,4 km por dia, o que é totalmente consistente com a taxa esperada de disseminação viral devido à dispersão de mosquitos silvestres e primatas, complementa Gonzalo. Dados obtidos por estudo da Superintendência de Controle de Endemias (Sucen), com base nas datas e localização das mortes dos macacos, teve resultados bastante próximos, apontando para uma velocidade de 2,3 km por dia nos meses mais quentes.
Uma análise futura, envolvendo genomas de regiões endêmicas e não endêmicas brasileiras, bem como de países vizinhos nos últimos anos, também seria crucial para entender melhor a evolução e a epidemiologia do vírus na América do Sul. Estudos baseados em abordagens genéticas reversas também serão importantes para determinar se as substituições de aminoácidos presentes na linhagem atual brasileira afetam a aptidão ou a transmissibilidade viral, explica Myrna Bolnado.
Para entender as mutações, modelos tridimensionais
Além da análise do genoma viral e das rotas de dispersão geográfica, o trabalho do grupo de pesquisadores teve ainda mais uma etapa. Para obter informações adicionais sobre a localização estrutural e os efeitos funcionais das substituições de aminoácidos nas proteínas NS3 e NS5, responsáveis pela replicação viral, pesquisadores do Laboratório de Genômica Funcional e Bioinformática do IOC construíram modelos tridimensionais baseados nas estruturas dessas proteínas. Esta etapa do estudo contou com a colaboração de especialistas do Programa de Computação Científica (PROCC) da Fiocruz.
|
Divulgação |
|
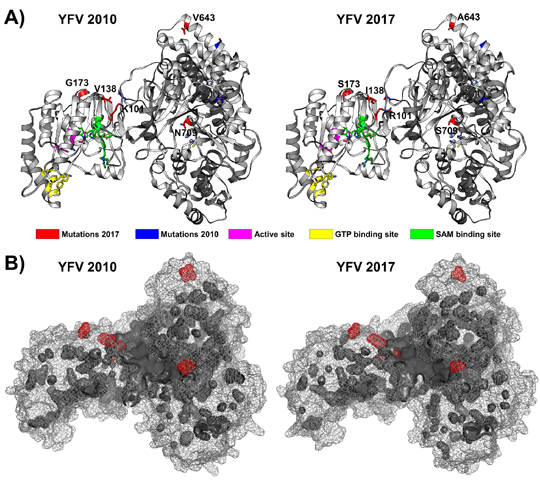
|
|
Modelo em três dimensões elaborado para o estudo: mapeando as alterações na proteína NS3 |
Para comparação, foram modeladas proteínas da linhagem brasileira atual e uma da Venezuela de 2010, depositada no banco internacional de genomas, considerada a ancestral mais recente sem as mutações descobertas a partir de 2017. Os cientistas identificaram que a maioria das mutações ocorre perto de regiões importantes para o desempenho das funções dessas proteínas, que podem ser afetadas por essas modificações.
Com esta parte da pesquisa foi possível analisar em detalhes de como as posições das novas substituições de aminoácidos podem implicar na estrutura da molécula, afetando atividades enzimáticas essenciais para a replicação viral. Há a hipótese de que as mudanças de aminoácidos tenham influenciado a capacidade de infecção viral nos homens, macacos e mosquitos, acelerando o surto, comenta a pesquisadora Ana Carolina Ramos Guimarães, do Laboratório de Genômica Funcional e Bioinformática, que atuou nas análises da modelagem 3D juntamente com Ernesto Raul Caffarena, do PROCC/Fiocruz e coordenador do programa de Pós-graduação em Biologia Computacional e Sistemas do IOC. No entanto, para avaliar o impacto funcional das mutações na atividade viral é necessário realizar testes adicionais, com experimentos em células e em animais chamados de testes in vitro e in vivo, respectivamente, completa.
As sequências genéticas completas dos vírus analisados no estudo já foram publicados no GenBank, de modo a estarem disponíveis para comparações que possam ser realizadas por outros cientistas do Brasil e do mundo. A disponibilização de dados é fundamental para que outros grupos, que também estudam o tema, possam produzir novas investigações, sintetiza a pesquisadora Ana Carolina Vicente, chefe do Laboratório de Genética Molecular de Microrganismos.
Reportagem: Vinicius Ferreira
Edição: Raquel Aguiar
20/12/2018
Permitida a reprodução sem fins lucrativos do texto desde que citada a fonte (Comunicação / Instituto Oswaldo Cruz


Instituto Oswaldo Cruz /IOC /FIOCRUZ - Av. Brasil, 4365 - Tel: (21) 2598-4220
| INTRANET IOC| EXPEDIENTE
Manguinhos - Rio de Janeiro - RJ - Brasil CEP: 21040-360
